

Abstract
Introduction: Myeloid neoplasms occurring after exposure to chemo- and/or radiotherapy, termed Therapy-related myeloid neoplasms (T-MN), are considered poor prognosis. This perception creates an inherent bias in treatment decision, resulting in either over- (allo-HSCT) or under-treatment (palliation) and exclusion from most clinical trials. The optimal T-MN treatment paradigm is unknown, partly because its mutational architecture is not well defined. Few small studies show mutations in only 50-60% (Ok, Leuk Res 2015; Lindsley, Blood 2015) of T-MN (compared to 80-90% in PMDS). This study compares the mutational architecture of T-MN and PMDS from the South Australian Myelodysplastic Syndrome registry.
Methods: Demographic, clinical and laboratory data including cytogenetic profiles of 129 T-MN (95 T-MDS; 34 T-AML [≥20% blasts]) and 108 PMDS patients were analysed. Targeted Massively Parallel Sequencing of a custom panel of 43 myeloid neoplasms associated genes (all coding regions) was performed on diagnosis bone marrow samples. Mutations with VAF ≥3% were selected for further assessment. Overall survival (OS) was calculated from date of diagnosis to date of last follow-up or death and adjusted using time varying covariate to account for disease modifying therapy (DMT) exposure.
Results: Compared to PMDS, T-MN patients were younger at diagnosis (71.1 vs 75.3 years, p<0.001), had higher frequency of IPSS-R High and Very High (VH) scores (39.5 vs 20.3%, p<0.001), poor risk cytogenetic abnormalities (del7/7q [18.4 vs 6.5%, p=0.0099], complex [28.8 vs 8.3%; p<0.0001] or monosomal karyotype [26.4 vs 4.6%; p<0.0001]), translocations (39.2 vs 10.2%; p<0.0001), and marker chromosomes (20.8 vs 1.9%; p<0.0001) and poor overall survival (10.6 vs 31.4 months, p<0.0001). Mutation frequency in our T-MN cohort was substantially higher than previously published frequency of 50-60% in t-MN and comparable to PMDS (93 vs 92.6%). The most common mutations were TP53 in T-MN and spliceosomal complex in PMDS. Within the spliceosomal complex, SF3B1 (25.9% vs. 6.2%; p<0.0001) and U2AF1 mutations (7.4% vs. 1.6%; p=0.04) were detected at higher frequency in PMDS compared to T-MN, while there was no significant difference in the SRSF2 (21.3% vs 15.5%) or ZRSR2 (5.6 vs 2.3%) mutations. Importantly, SRSF2 mutations were the most frequent spliceosome mutations in T-MN, in contrast to SF3B1 in PMDS.
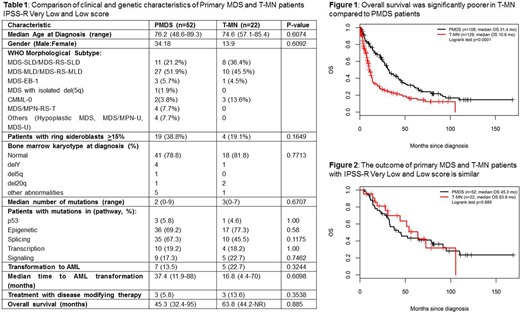
Although the OS was poorer for T-MN than PMDS (10.6 vs 31.4 mo, p<0.0001, Figure 1) as a group, the characteristics of T-MN and PMDS were different when segregated according to Revised International Prognostic Scoring System (IPSS-R) score. T-MN with intermediate IPSS-R score had poorer OS compared to PMDS (13.6 vs 35 mo, p=0.01). T-MN with High or VH IPSS-R scores had poorer OS than PMDS (8.8 vs 12.8 mo) but was not significant (p=0.09) due to application of DMT in a significant subset of patients. Interestingly, survival of T-MN with VL or Low risk was similar to PMDS (63.8 vs 45.3 mo, p=0.9, Figure 2). Hence, we compared the clinical and genetic characteristics of PMDS (n=52) and T-MN (n=22) patients with VL or Low IPSS-R score (Table 1). There was no significant difference between their demographics and MDS subtypes. A significant majority of patients had normal karyotype; no poor-risk chromosomal abnormalities were seen in remainder. The mutation profile of both groups was also remarkably similar. The median number of mutations and frequency of mutated genes were not different.
As expected, VL or Low risk T-MN patients were different from T-MN with intermediate, High or VH IPSS-R score (n=74) who showed higher frequency of high-risk karyotypes and poorer OS. Additionally, the mutation profile was also different between the two groups; TP53 mutations were less common while TET2, SF3B1, IDH2 mutations were significantly more frequent in low-risk T-MN.
Conclusions: In our cohort, somatic mutations were seen in 93% of T-MN which is higher than published literature. TP53 and spliceosomal mutations were commonest in T-MN and PMDS, respectively. Although, the T-MN survival is poorer than PMDS, the subgroup with IPSS-R Very Low or Low risk mirrors clinical and genetic characteristics of PMDS and has similar outcome. Such patients should be managed at par with PMDS counterparts and, importantly, should not be excluded from appropriate clinical trials based on pre-MDS chemotherapy or radiotherapy exposure.
Hiwase:Novartis: Research Funding; Celgene: Research Funding.
This icon denotes a clinically relevant abstract

